Última actualizacón: 22 octubre 2018 a las 11:18
Los lectores de este blog ya sabréis que tengo un especial interés en conocer todos los avances que se producen en relación con la fiebre mediterránea familiar (FMF). Esta enfermedad es un desorden autoinflamatorio sistémico caracterizado por episodios recurrentes de fiebre, serositis 1, sinovitis 2 y, en algunos casos, por inflamaciones cutáneas. Esta inflamación viene acompañada generalmente de una crisis de dolor abdominal autolimitada.
Pues bien, hoy voy a analizar un interesantísimo trabajo publicado hace escasos días en la prestigiosa revista Expert Opinion on Biological Therapy y que aporta novedades importantes en los tratamientos disponibles para la enfermedad.
Hasta hace poco tiempo, el único tratamiento para la FMF consistía en la toma de un comprimido: colchicina. La colchicina se comercializa en España bajo la presentación Colchimax (que une a la colchicina la dicicloverina hidrocloruro, un antidiarréico que trata de contrarrestar el principal efecto secundario). La dosis normal es de 1,2 a 2,4 mg al día en adultos, pudiendo incrementarse o reducirse en 0,3 mg al día en función de la eficacia o los efectos secundarios. La toma de este medicamento es para toda la vida ya que no solo controla los brotes de la enfermedad sino que elimina el riesgo de sufrir amiloidosis, la manifestación más grave que, en ocasiones, puede llevar a la muerte del enfermo. Debemos añadir que la seguridad y eficacia de este fármaco es muy alta.
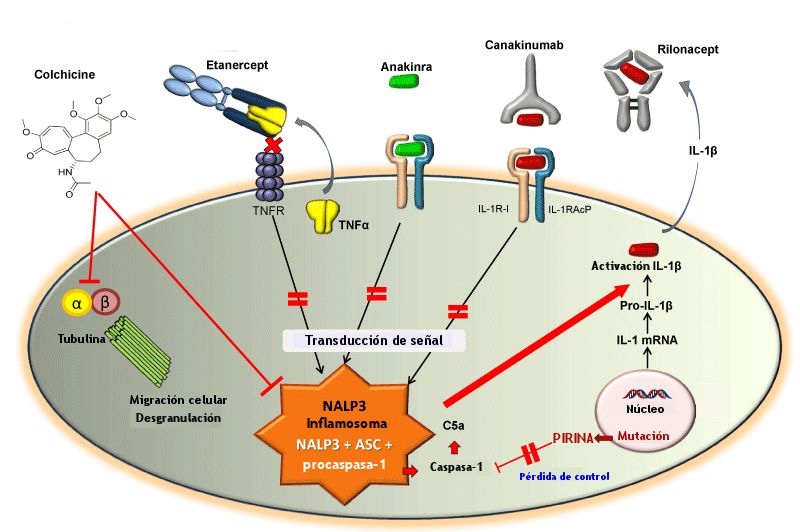
Sin embargo, se da la circunstancia de que entre un 5 y un 10% de los pacientes con FMF son resistentes –no responden bien– al tratamiento con colchicina; resistencia que se define clínicamente cuando se sufre al menos una crisis al mes a pesar de mantener un tratamiento diario con 2 mg o más del fármaco. Existen varias hipótesis acerca del mecanismo de acción de la colchicina y recientemente se ha demostrado que suprime la activación de la caspasa-1, bloqueando la conversión de pro-IL-1β (proteínas precursoras activas de la interleucina-1β) en IL-1β. 3.
Durante décadas éste fue el único tratamiento conocido, pero los descubrimientos relacionados con la principal causa de la enfermedad, la alteración del inflamosoma, así como el papel de la pirina en la regulación de la activación de la IL-1β como la citocina más importante involucrada en la patogénesis de la FMF, llevaron a los investigadores a plantear la posibilidad de bloquearla mediante un antagonista.
Actualmente disponemos de tres tratamientos biológicos que actúan como antagonistas para la IL-1β: anakinra, rilonacept y canakinumab. Estos medicamentos son agentes inmunosopresores cuya función es la de bloquear la actividad biológica de la IL-1β producida naturalmente, con lo que ataca la base de la reacción inflamatoria.
Distintos tratamientos para la FMF
Hasta la fecha se han realizado pocos ensayos controlados acerca de la eficacia de estos tratamientos 4. Y comprobar el estado de la cuestión ha sido la labor que han emprendido los Doctores Haviv y Hashkes que han realizado una búsqueda bibliográfica de tratamientos donde se han empleado antagonistas de la IL-1β en enfermos con FMF –en particular de canakinumab, un anticuerpo humanizado de la IL-1β– mediante una búsqueda en tres bases de datos científicas (PubMed, Medline y Scopus) desde el año 2001 (cuando comenzó la comercialización de anakinra); así como en las actas de los más importantes congresos reumatológicos desde 2011 (para contabilizar los estudios no publicados aún).
Pero antes de entrar en detalles y para comprender en profundidad lo que vamos a exponer, tenemos que saber qué es un ensayo clínico, qué es un protocolo así como otros aspectos relevantes de la investigación sobre nuevos tratamientos para las enfermedades.
Según el artículo 2 del Real Decreto 223/2004, de 6 de febrero, por el que se regulan los ensayos clínicos con medicamentos, un ensayo clínico es «toda investigación efectuada en seres humanos para determinar o confirmar los efectos clínicos, farmacológicos y/o demás efectos farmacodinámicos, y/o de detectar las reacciones adversas, y/o de estudiar la absorción, distribución, metabolismo y excreción de uno o varios medicamentos en investigación con el fin de determinar su seguridad y/o su eficacia». Más allá de los defectos gramaticales de esta definición, vemos que un ensayo clínico es una forma de obtener datos casi reales en pacientes casi reales acerca de la forma de actuar de un determinado medicamento, con el objetivo de interpretarlos y respaldar o no la continuidad del plan de investigación.
Para ello, se hace preciso contar con un protocolo, que es el «documento donde se describen los objetivos, el diseño, la metodología, las consideraciones estadísticas y la organización de un ensayo».
En este sentido, conviene saber que hay diferentes tipos de estudios (sólo mencionaremos los relevantes a efectos de esta anotación):
- Estudios de casos. Los casos (pacientes con una determinada enfermedad) son seleccionados por el investigador entre un grupo de pacientes disponibles. Además de este grupo, se puede seleccionar un segundo (o más) grupo de individuos sanos que servirán como controles. Una vez identificados los casos y los controles, se miden los resultados buscados de forma retrospectiva.
- Ensayo controlado. En un ensayo controlado, la exposición al fármaco bajo evaluación se compara frente a un grupo de control que recibe un placebo. La asignación de los pacientes a uno u otro grupo se hace de forma aleatoria, es decir, los pacientes se distribuyen u organizan al azar. Destacamos aquí el término inglés double-blind que, siguiendo a Pablo Mugüerza 5, traducimos como «enmascaramiento doble» mejor que la más extendida «doble ciego»: Se trata de ensayos donde ni los pacientes ni el investigador conocen el tratamiento que se administra a cada grupo. Es decir, el elenco de participantes sabe que existe un fármaco y un placebo, pero lo que no sabe ninguno es a qué paciente se ha asignado cada cual. Decir también que el ensayo clínico ideal incluye al menos dos grupos de pacientes. Uno recibe el tratamiento evaluado (es decir, el grupo experimental), y el otro recibe un placebo u otro tratamiento con el que se pretende comparar el primero (grupo de referencia).
Con lo dicho hasta ahora, podemos comprender que el diseño más apropiado para establecer una relación de causalidad entre un medicamento y un efecto positivo (o negativo) para el tratamiento de una enfermedad es el de ensayo controlado que disponga de un protocolo bien formulado.
Por último, conviene tener en cuenta que un ensayo clínico se divide en diferentes fases o estadios 6:
- Fase I: Constituye el primer paso en la investigación de una sustancia o medicamento nuevo en el hombre. Son estudios que proporcionan información preliminar sobre el efecto y la seguridad del medicamento en sujetos sanos o en algunos casos en pacientes, y servirán para establecer la pauta de administración más apropiada para ensayos posteriores.
- Fase II: Se realiza en pacientes que padecen la enfermedad o entidad clínica de interés. Tiene como objetivo proporcionar información preliminar sobre la eficacia del fármaco, establecer la relación dosis-respuesta del mismo, conocer las variables empleadas para medir eficacia y ampliar los datos de seguridad obtenidos en la fase I. Por lo general, estos ensayos clínicos serán controlados y con asignación aleatoria de los tratamientos.
- Fase III: Son ensayos destinados a evaluar la eficacia y seguridad del tratamiento experimental intentando reproducir las condiciones de uso habituales y considerando las alternativas terapéuticas disponibles en la indicación estudiada. Se realiza con una muestra de pacientes más amplia que en la fase anterior y representativa de la población general a la que irá destinado el medicamento.
- Fase IV: Son ensayos clínicos que se realizan con un medicamento después de su comercialización. Podrán ser similares a los descritos en las fases I, II, III si estudian algún aspecto aún no valorado o bajo condiciones de uso distintas de las autorizadas (por ejemplo, una nueva indicación).

Los resultados del primer estudio de casos en el que se utilizó un antagonista de la IL-1β como terapia para la FMF se publicaron en 2007: se empleó anakinra para tratar un paciente resistente a la colchicina. La primera publicación de un estudio controlado iba referida al uso de rilonacept, un receptor soluble de la IL-1β que la «atrapa» impidiendo su actuación.
En la mayoría de los casos, la administración de anakinra tuvo un efecto dramático en cuestión de horas o días, mientras que las proteínas de fase aguda se normalizaron en una semana. Sin embargo, no todos los pacientes respondieron por completo. Un estudio francés denominado MAIL1 (Maladies Auto-inflammatoires et Anti-IL-1) expuso una tasa de respuesta parcial del 46,2%.
Por su parte, el equipo del Dr. Ben-Zvi ha realizado un ensayo cuyos resultados aún no se han publicado (fue comunicado este mismo año 2016 en un congreso de la especialidad). Se trata de un ensayo controlado de cuatro meses de duración, con enmascaramiento doble, aleatorizado y comparativo con placebo, donde participaron 25 pacientes de FMF resistentes a la colchicina. Los criterios de inclusión fueron los siguientes: pacientes adultos FMF (con edades comprendidas entre los 18 y los 65 años), que cumplieran con los criterios de Tel-Hashomer para el diagnóstico de FMF, con dos mutaciones en el gen MEFV, y que sufrieran al menos un ataque febril al mes a pesar de recibir ≥2 mg al día de colchicina.
Doce pacientes recibieron anakinra (100 mg al día) y el resto recibieron un placebo. La tasa de ataques, el resultado principal buscado con el ensayo, fue significativamente menor en el grupo experimental (cuyos miembros recibieron el medicamento) que en el de referencia (1,7 ± 1,7 frente a 3,5 ± 1,9 ataques por mes y paciente, respectivamente). Siete pacientes abandonaron el estudio antes de tiempo (todos recibieron placebo). La calidad de vida fue significativamente mejor en el grupo experimental y no se registraron eventos adversos graves.
En lo tocante al canakinumab, en 2011 se publicaron los primeros estudios de casos (case reports en inglés) acerca de su utilidad como tratamiento para la FMF. Acto seguido se pasó a ensayos en fase II (anunciados en 2014 y 2015 y con dos grupos, uno de niños y otro de adultos) cuyo éxito permitió pasar a un ensayo en fase III: un ensayo con enmascaramiento doble, aleatorizado y comparativo con placebo en pacientes con FMF resistentes a la colchicina.
El canakinumab como tratamiento para la FMF
Canakinumab (ACZ885, Ilaris®; Novartis Pharma, Basilea, Suiza) es un anticuerpo monoclonal recombinante humanizado 7 que actúa como antagonista de la IL-1β. Este fármaco ha sido aprobado por la Administración de Medicamentos y Alimentos estadounidense (FDA) y la Agencia Europea del Medicamento (EMA) para el tratamiento de los síndromes periódicos asociados a la criopirina (CAPS) y la artritis idiopática juvenil sistémica. También ha sido aprobado por la EMA para tratar los síntomas de la artritis gotosa, cuando otros fármacos no son tolerados, están contraindicados o no tienen un efecto adecuado.
Analizamos a continuación la situación clínica de este medicamento:
Estudio de casos y series de casos: El canakinumab ha sido una opción de tratamiento eficaz para la gran mayoría de los pacientes tratados, aunque algunos pacientes sólo tuvieron una respuesta parcial, sobre todo cuando se administró con una frecuencia inferior a una inyección cada cuatro semanas.
Ensayo en fase II 8: Se trató de ensayos de seis meses de duración, sin enmascaramiento y con un solo grupo. Los criterios de inclusión fueron los siguientes: diagnóstico de FMF de acuerdo con los criterios de Tel-Hashomer; dos mutaciones documentadas en el exón 10 del gen MEFV (salvo dos pacientes heterocigotos que fueron admitidos en la cohorte de Turquía) y ataques con una frecuencia media de ≥1 por mes durante los tres meses anteriores a la selección, a pesar de seguir un tratamiento con ≥1-2 mg al día de colchicina, durante al menos tres meses. Tras un seguimiento, los participantes que sufrieron uno o más ataques de FMF confirmados por el investigador durante los 30 días del periodo de experimentación, fueron seleccionados para recibir el tratamiento, que consistió en tres inyecciones subcutáneas de canakinumab (2 mg/kg, máximo 150 mg) cada cuatro semanas, mientras mantenían su tratamiento con colchicina de forma habitual.
Ensayo en fase III: El diseño y los resultados de los ensayos en fase III no se han publicado aún de forma completa, de ahí que sólo podamos conocer aquellos resultados cualitativos publicados en forma de resumen o póster en los congresos de la especialidad.
Este estudio internacional con enmascaramiento doble, aleatorizado y comparativo con placebo ha logrado inscribir a más de 200 participantes, aunque bajo un protocolo general para tres enfermedades: Deficiencia de Mevalonato Quinasa, Síndrome periódico asociado al receptor 1 del factor de necrosis tumoral (TRAPS) y enfermos de FMF resistentes a la colchicina. Los criterios clave de elegibilidad en relación con la FMF eran: pacientes con dos o más años de edad, con al menos una mutación en el exón 10 del gen MEFV, y con crisis periódicas a pesar del tratamiento con colchicina.
El estudio se dividió en 4 periodos:
- Un período de cribado o selección de hasta 12 semanas.
- Un periodo fundamental aleatorizado, con enmascaramiento doble, y comparativo con placebo, con grupos en paralelo 9 de 16 semanas de duración. Los pacientes se asignaron al azar 1:1 para recibir 150 mg canakinumab 10 o un placebo cada cuatro semanas.
- Un periodo de retirada aleatorizada del tratamiento para los pacientes que respondían bien al mismo de 24 semanas.
- Un periodo de extensión del tratamiento sin enmascaramiento.
El objetivo principal del ensayo fue comparar los grupos que habían recibido canakinumab y aquellos que recibieron placebo para obtener la proporción de participantes que habían dejado de tener crisis dentro de los 15 días desde el inicio del tratamiento, así como nuevas crisis en las 16 semanas siguientes. Los objetivos secundarios incluían la proporción de pacientes con brotes mínimos en la semana 16, la proporción de pacientes con niveles de proteína C reactiva y niveles de proteína amiloide sérica normales en la semana 16 11. La calidad de vida relacionada con la salud se evaluó utilizando herramientas similares a los ensayos en fase II.
Sólo disponemos de los resultados del segundo periodo donde, de manera significativa, hubo una proporción mayor de pacientes tratados con canakinumab que alcanzaron el objetivo principal y se consideró que respondían bien al tratamiento frente a los que recibieron placebo. También se han alcanzado todos los objetivos secundarios.
No se han descrito efectos adversos entre los pacientes de FMF tratados con canakinumab, aunque debemos tener presente que el número de pacientes analizados es aún pequeño, y el nivel de seguimiento en el tiempo relativamente corto. Los acontecimientos adversos más frecuentes fueron nasofaringitis (resfriados) y síntomas gastrointestinales. Otros efectos secundarios comunes incluyen gripe y dolor de cabeza.

Conclusiones
De los tres medicamentos anti-IL-1β disponibles en el mercado (anakinra, rilonacept y canakinumab), el más cómodo para el paciente es canakinumab debido a su larga semivida y la facilidad de uso (recordemos que basta una inyección subcutánea cada cuatro semanas aproximadamente). Esta cuestión es especialmente relevante para una enfermedad genética que puede necesitar tratamiento de por vida.
En cualquier caso, es necesario definir la dosis y el intervalo de administración óptimos. En aquellos pacientes que no responden al tratamiento, o no lo hacen del todo, es posible que un aumento de la dosis o el cambio a otro inhibidor de IL-1β sea una estrategia eficaz a pesar de que los estudios controlados publicados hasta la fecha no abordan adecuadamente esta cuestión. Del mismo modo, tampoco se analiza cuánto tiempo debe durar el tratamiento (se supone que es de por vida como hemos dicho) y cómo retirarlo.
Por último, otra cuestión importante que debe abordarse es si durante el tratamiento con inhibidores de la IL-1β debe mantenerse el tratamiento con colchicina. En la actualidad existe un consenso de que sí es necesario mantener la colchicina, ya que es el único fármaco que ha demostrado prevenir la amiloidosis. Sin embargo, es posible que se pueda administrar una dosis más baja, todo ello a partir de valoraciones periódicas del estado del paciente.
![]()
Referencias
Haviv, R. y Hashkes, P. J. (2016), «Canakinumab investigated for treating familial Mediterranean fever«. Expert Opinion on Biological Therapy, vol. 16, núm. 11, p. 1425-1434.
Mugüerza, P. (2012), Manual de traducción inglés-español de protocolos de ensayos clínicos. Cuadernos de la Fundación Dr. Antonio Esteve, Nº 23. Barcelona: Fundación Dr. Antonio Esteve, 184 p.
Navarro González, F. (2005), Diccionario crítico de dudas inglés-español de medicina. Madrid: McGraw-Hill, 1133 p.
Saladrigas, M. V., et al. (2008), «Glosario EN-ES de ensayos clínicos (1ª parte: A-M)». Panace@, vol. 9, núm. 27, p. 8-54.
Saladrigas, M. V., et al. (2008), «Glosario EN-ES de ensayos clínicos (2ª parte: N-Z)». Panace@, vol. 9, núm. 28, p. 107-141.
Más información en la página de la Asociación Española de Fiebre Mediterránea Familiar STOP-FMF.
Notas
- Inflamación de los tejidos serosos del cuerpo: como los tejidos que rodean los pulmones (pleura), el corazón (pericardio), y la capa interior del abdomen (peritoneo). ↩
- Cualquier inflamación a nivel de las estructuras que revisten las articulaciones. ↩
- Ver Slobodnick, A., et al. (2015), «Colchicine: Old and new«. The American Journal of Medicine, vol. 128, núm. 5, p. 461-470. ↩
- A excepción del rilonacept, donde se ha comprobado su eficacia en un grupo de 12 pacientes de FMF resistentes a la colchicina aunque, como vemos, en una muestra muy reducida ↩
- Ver Mugüerza, P. (2012), Manual de traducción inglés-español de protocolos de ensayos clínicos. Cuadernos de la Fundación Dr. Antonio Esteve, Nº 23. Barcelona: Fundación Dr. Antonio Esteve, 184 p. ↩
- Adaptado de la página de la Universidad de Barcelona ↩
- Que contiene un 90% de material humano, lo que reduce la inmunogenicidad de los anticuerpos, es decir, el rechazo del sistema inmunitario ↩
- Ensayos de viabilidad, donde se evalúa qué dosis seguirá adelante con el proceso de análisis, y también la eficacia de esta dosis ↩
- Donde la medida diagnóstica objeto de estudio y la medida de referencia se asignan, mediante una técnica de aleatorización, a dos o más grupos de sujetos o pacientes, y cuya evolución se sigue de forma paralela a lo largo del estudio ↩
- O 2 mg/kg para pacientes con un peso ≤40 kg ↩
- La proteína C reactiva y la proteína amiloide sérica son marcadores habituales para la inflamación sistémica ↩