Última actualizacón: 2 marzo 2018 a las 19:33
Esta anotación es una ampliación de otra que publiqué hace unos meses con ocasión del día mundial de las enfermedades raras (titulada Yo también soy raro) con la finalidad de explicar qué es la fiebre mediterránea familiar (apoyándome en mi experiencia personal) así como analizar las últimas investigaciones en este campo. En estos momentos participo como ponente en el evento de divulgación científica Desgranando Ciencia dando una charla sobre este particular pero, debido a las restricciones de tiempo, he creído oportuno ofrecer aquí una información más detallada así como enlaces a los artículos científicos que menciono.
La fiebre mediterránea familiar (en adelante FMF) es un desorden autoinflamatorio sistémico caracterizado por episodios recurrentes de fiebre, serositis , sinovitis y, en algunos casos, por inflamaciones cutáneas. Esta inflamación viene acompañada generalmente de una crisis de dolor abdominal autolimitada. La enfermedad se hereda de forma autosómica recesiva, lo que significa que se manifiesta únicamente cuando la mutación del gen MEFV que la provoca está presente en las dos copias del material genético del enfermo. Para comprender esto debemos saber que en cada una de nuestras células existen dos copias completas de nuestro ADN: una la recibimos de nuestro padre y otra copia de nuestra madre.
En el caso de la FMF, el que se trate de una enfermedad recesiva implica que para que se manifiesten los síntomas tienen que darse las dos copias mutadas del gen MEFV ya que, en caso contrario, la copia “buena” del gen supliría las deficiencias del otro.

Gen autosómico recesivo. Modo de herencia entre dos portadores de un gen autosómico recesivo (wikimedia commons)
Para que se den cuenta de la casualidad (y como refleja perfectamente la imagen de más arriba), se ha dado la circunstancia de que cada uno de mis padres tiene una copia mutada del gen MEFV (así, son portadores de la enfermedad y pueden transmitirla pero no la padecen) y debido a la recombinación genética, esas dos copias vinieron a juntarse en mí (de hecho, mis hermanos también pueden ser portadores aunque ninguno de ellos la sufre). Por esto la “familiaridad” de la enfermedad se manifiesta no tanto en familiares directos (padres, hermanos o abuelos) como en primos o tíos.
¿Cuáles son los síntomas?
Como ya hemos adelantado, en algunos casos la fiebre puede ser el único síntoma con temperaturas que oscilan entre los 38 y los 40 ºC.
Sin embargo, en la mayoría de los pacientes la fiebre sólo es la antesala de la manifestación del resto de síntomas, donde el ataque de dolor abdominal es el más habitual ya que lo sufren el 95% de los mismos. Este dolor suele estar localizado al principio de la fase aguda (generalmente en la fosa ilíaca derecha) para extenderse por el resto del abdomen con el paso de las horas. La localización anatómica del dolor y la forma de presentarse junto con la fiebre llevan en muchas ocasiones a que los servicios de urgencias hospitalarias decidan realizar laparotomías o apendicetomías que, a la postre, se revelan innecesarias.
La artritis, o dolor articular, es otra característica común de esta enfermedad, viéndose más afectadas las articulaciones de las extremidades inferiores.
Por último, un tercio de los pacientes manifiestan una inflamación de los tejidos que rodean los pulmones (pleuritis) que desembocan en ataques agudos de fiebre y dolor de pecho.
Debemos tener presente que la casuística es enormemente variada ya que los enfermos no tienen por qué experimentar todos los síntomas. A esta circunstancia se une el hecho de que la enfermedad varía en su manifestación a lo largo de la vida de los pacientes, quienes experimentan diferentes tipos de crisis. De hecho, aunque la edad habitual de aparición de los síntomas es alrededor de los 20 años , también se han descrito casos durante la lactancia y la niñez, con la enorme dificultad diagnóstica que eso supone.
Por último destacar que la complicación más grave e importante es el desarrollo de amiloidosis. El amiloide es un material de naturaleza proteica que se deposita en ciertos órganos (riñones, hígado, intestino, piel y corazón) causando una pérdida progresiva de su función. La amiloidosis no es una complicación exclusiva de la FMF ya que puede aparecer en el curso de cualquier enfermedad inflamatoria crónica que no se trate adecuadamente y puede, en los casos más severos, llevar a la muerte.
En mi caso particular ¿cómo se manifiesta la enfermedad?
Mi principal síntoma es un fuerte dolor abdominal que comienza en la fosa ilíaca derecha y termina extendiéndose por toda la región abdominal, acompañado de fiebre alta (picos de 39º y 40º). Además tengo artritis en las articulaciones, sobre todo en las muñecas y las rodillas (ya cronificada aún sin crisis). Esta fase aguda o “crisis” tiene una duración variable que va desde las 24 horas hasta los dos o tres días. La evolución de las crisis es progresiva y autolimitada: comienza con una leve molestia abdominal muy focalizada que poco a poco se va intensificando y generalizando hasta que alcanza su máxima intensidad unas cuatro o cinco horas más tarde. Esta situación llega a desaparecer por completo al cabo de pocos días .
Además de la duración, otro aspecto importante es la frecuencia, la recurrencia de las crisis. En este aspecto también hay una gran variabilidad ya que hay épocas en las que sufro una crisis al mes, y otras en las que me libro durante 3 o 4 meses. Esta ha sido la tendencia habitual durante muchos años.
Sin embargo, de un tiempo a esta parte se ha venido a complicar la cosa. No sólo ha aumentado la frecuencia de las crisis (ahora tengo una crisis cada semana o 10 días, lo que en la práctica supone que estoy enfermo todo el tiempo) sino que se ha sumado la pleuritis a los síntomas, que quizás sea la manifestación más molesta. Se trata de ataques agudos de fiebre y dolor de pecho que comienzan justo cuando pasa el dolor abdominal y duran otras 24 o 48 horas en plenitud.
El sistema inmunitario
Para entender el mecanismo que desencadena la enfermedad necesitamos tener claros algunos conceptos de inmunología. La inmunidad es la capacidad del organismo para defenderse contra cualquier elemento extraño. Por lo tanto, el sistema inmunitario tiene como primera función reconocer lo propio de lo ajeno para, una vez reconocido lo ajeno, desencadenar una serie de mecanismos que conducirán a su eliminación . De esta forma, una de las tareas esenciales del sistema inmunitario es la de aprender a reconocer sus propias estructuras: este proceso se denomina tolerancia inmunológica.
Se trata de una cuestión muy importante ya que un exceso de tolerancia hará que el organismo no reconozca todas las sustancias extrañas que debiera, tornándose incapaz de defenderse (inmunodeficiencia); mientras que una falta de tolerancia provocará que reconozca como extrañas las propias estructuras del organismo, dando origen a la autoinmunidad.
Siguiendo esta argumentación, podemos dividir el sistema inmunitario en función de que los elementos del sistema requieran reconocer previamente al agente extraño (la llamada inmunidad específica, adaptativa o adquirida ) o no reconocerlo (inmunidad natural, innata o inespecífica ) para poder actuar. Veamos algunas pinceladas de cada uno:
Si un microorganismo o agente extraño logran atravesar la piel y los epitelios se pone en marcha el sistema de inmunidad natural. Se trata de una red de células y proteínas que responden a la infección o a la lesión de los tejidos a través del reconocimiento genéticamente «programado» de las moléculas extrañas (por ejemplo, los componentes de la pared celular bacteriana) o moléculas producidas o liberadas por las células dañadas (por ejemplo, la interleucina 1 (IL-1) y los cristales de ácido úrico).
Por otro lado, el sistema de inmunidad específica constituye una barrera defensiva adicional, aún más sofisticada, formado por un tipo de moléculas que funcionan como «adaptadores flexibles» llamados anticuerpos, que por un lado se unen a los fagocitos , y por el otro se unen al microorganismo (llamado antígeno) sin importar de qué tipo se trate. De esta forma los atrapan y eliminan.
La inmunidad específica se caracteriza por dos hechos esenciales: primero, hay una memoria de cada encuentro con el antígeno, lo que hace que cada contacto posterior amplifique la respuesta (esta es la base de la vacunación); y segundo, la respuesta inmunitaria específica amplifica los mecanismos protectores de la inmunidad natural, logrando que los antígenos sean localizados y eliminados más rápidamente.
Otro componente fundamental del mecanismo de respuesta inmunitaria es el llamado sistema del complemento. El sistema del complemento es un sistema funcional de proteínas del plasma (cerca de 40) que interactúan entre sí y con otros elementos de los sistemas inmunitarios innato y adquirido, para mediar una serie de respuestas inmunológicas. La activación del sistema del complemento induce la producción de péptidos que activan, entre otras, la respuesta inflamatoria dando como resultado un flujo de fluidos que lleva anticuerpos y células fagocitarias al lugar de entrada del antígeno.
Las enfermedades autoinflamatorias, como la FMF, surgen a consecuencia de la activación inapropiada de los mecanismos inflamatorios independientes de antígenos, es decir, del sistema de inmunidad natural. Lo analizaremos con más detalle a continuación.
Genética
Se ha identificado la mutación responsable de la FMF en un gen, llamado MEFV, que está compuesto por 10 exones localizados en el brazo corto del cromosoma 16 (16p13.3). De acuerdo con la base de datos Infevers, a día de hoy se han estudiado 298 variaciones asociadas a la enfermedad, la mayor parte de las cuales están agrupadas en el último exón (exón 10), aunque la mutación localizada en el exón 2 (E148q) parece ser la más común en los enfermos europeos .

Esquema del gen MEFV. Infevers
Este gen codifica para una proteína formada por 781 aminoácidos llamada pirina o marenostrina que participa en el control de la inflamación, por lo que si está mutado, como sucede en los enfermos de FMF, esta regulación no se lleva a cabo correctamente.
La pirina tiene un papel fundamental en el sistema inmune innato, y se expresa en el citoplasma de ciertos leucocitos y también se localiza en los microtúbulos. Su principal función es la de regular el inflamosoma, una estructura conformada por proteínas intracelulares implicadas en el inicio de la respuesta inflamatoria, necesario para la conversión de pro-interleucina 1β a interleucina 1β activada.
¿Qué pasa concretamente con este gen MEFV?
La respuesta típica de la inmunidad natural frente a una agresión externa es la inflamación. Si se produce una rotura en la piel los microorganismos pueden acceder al interior del organismo de ahí que, como reacción, se produzca la inflamación con varios fines: aislar, destruir al agente dañino, y reparar el tejido u órgano dañado. Todos conocemos los signos: rubor (coloración roja), tumor (hinchazón), calor y dolor. La coloración y el calor se deben a un aumento del flujo sanguíneo en el área y a la constricción de los vasos sanguíneos. Gracias a la intervención de mediadores químicos se aumenta la permeabilidad capilar para que los líquidos y las células sanguíneas pasen al espacio extravascular: esto provoca la hinchazón y el aumento de la presión local, que es lo que origina el dolor. El objeto de todo este movimiento orquestal es la acumulación de un fluido rico en proteínas, fibrina y leucocitos. Éstos últimos se dirigen hacia la zona afectada por un proceso denominado quimiotaxis (una locomoción dirigida) para, una vez allí, fagocitar los microbios y destruirlos.

Respuesta inflamatoria.
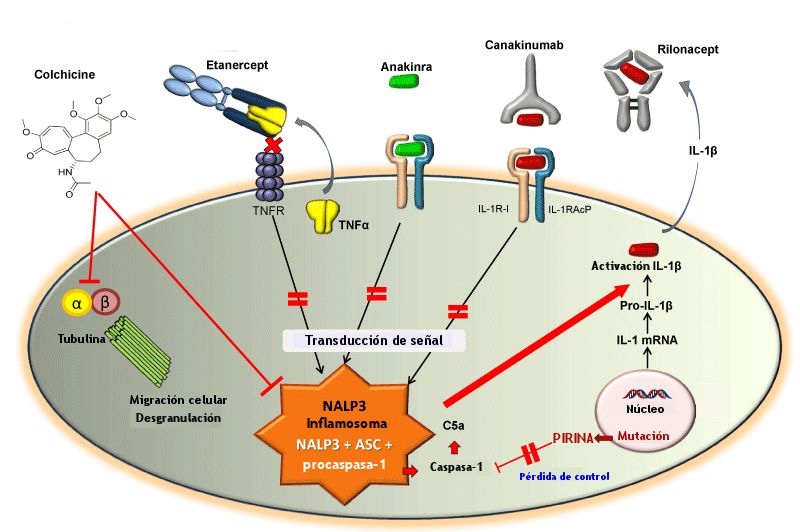
La desregulación del inflamosoma se ha propuesto como el principal mecanismo de la FMF. Entre las sustancias que intervienen en la quimiotaxis se encuentran los componentes del sistema del complemento, sobre todo la proteína C5a. Para que nos entendamos, cada vez que se produce una lesión, la C5a se libera con la función de decirle a los neutrófilos (que son quienes tienen que combatir la infección) ven aquí y defiende el organismo. Son los primeros en llegar al lugar dañado y, además de trabajar como basureros, engullendo literalmente los agentes nocivos, actúan como mediadores de la inflamación (la causan y, al mismo tiempo, cuando la necesidad de la misma ha desaparecido, se encargan de su desaparición).
En las personas sanas, la pirina inhibe el mediador de la inflamación C5a, lo que sirve para controlar la presencia de neutrófilos y, por tanto, la inflamación. Sin embargo, en una persona con FMF la ausencia de función de la pirina debido a la mutación del gen MEFV lleva a una sobre secreción de citocinas inflamatorias . El resultado es una espiral ascendente que culmina en una reacción inflamatoria desmedida: el ataque de FMF.
A un nivel clínico, los ataques agudos de FMF se producen debido a la activación de los neutrófilos en las superficies serosas y sinoviales. El papel clave de los neutrófilos en la FMF se ha confirmado gracias a diversas circunstancias: están presentes en el fluido seroso; la pirina se expresa en los neutrófilos; y porque la colchicina tiene un efecto rápido en la prevención de los ataques.

Adaptado de Wang, D., et al. (2014), «Familial Mediterranean Fever: From pathogenesis to treatment». Journal of Genetic Syndromes & Gene Therapy, vol. 5, núm. 5, p. 1-11.
¿Cuál es el tratamiento?
El tratamiento consiste en la toma de un medicamento: colchicina. La colchicina se comercializa en España bajo dos presentaciones: Colchicina Seid y Colchimax (que une a la colchicina la dicicloverina hidrocloruro, un antidiarréico). La dosis normal es de 1,2 a 2,4 mg al día en adultos, pudiendo incrementarse o reducirse en 0,3 mg al día en función de la eficacia o los efectos secundarios.
La toma de este medicamento es para toda la vida ya que no solo controla los brotes de la enfermedad sino que elimina el riesgo de sufrir amiloidosis.
Me voy a detener un momento para explicar cómo se llegó a utilizar la colchicina para tratar la FMF. Tenemos que situarnos en el Boston de la década de los sesenta del siglo pasado. En el Hospital General de Massachusetts, el Dr. Stephen Goldfinger atendió a una paciente que le había remitido un colega psiquiatra: una mujer con una profunda depresión debido a que sufría crisis recurrentes de dolor abdominal y fiebre que no le permitían llevar una vida normal. Como todos los tratamientos habían fallado, el Dr. Goldfinger le dijo que no podía ofrecerle otra cosa que narcóticos para mitigar en lo posible los dolores. Días más tarde, mientras estaba en un congreso fuera de la ciudad, el médico recibió un mensaje urgente ya que su paciente había intentado suicidarse debido a la desesperación (pasó cuatro días en coma aunque al final sobrevivió).
Impactado por la situación, el Dr. Goldfinger decidió implicarse a fondo para encontrar un tratamiento efectivo para su paciente. Consultó a varios de sus colegas sin obtener resultados hasta que un día, mientras comía en la cafetería del hospital, volvió a preguntar si alguien conocía algún tratamiento para la FMF o tenía alguna pista que ofrecer sobre el caso. Para su sorpresa recibió una respuesta afirmativa del Dr. Guillermo Sánchez: le explicó que tenía un paciente de gota que también padecía FMF y que cuando le prescribió colchicina (el medicamento habitual para tratar los ataques de gota), los ataques de FMF desparecieron.
Sin pensarlo dos veces, el Dr. Goldfinger comenzó a tratar a su joven paciente con colchicina a diario y el resultado fue casi inmediato: no volvió a tener más ataques.
Tras este éxito inicial, comenzó un estudio con cinco pacientes de FMF a quienes administró entre 0,6 y 1,8 mg de colchicina al día. Los resultados, que corroboraban el descubrimiento, fueron rechazados por la revista New England Journal of Medicine (NEJM) debido a que no cumplían el estándar científico de la revista y no se habían llevado a cabo ensayos de doble ciego. A pesar de todo se aceptó su publicación en forma de “carta” a la revista (que exigía un baremo de rigor inferior) y el artículo vio la luz en 1972 bajo el título Colchicine for Familial Mediterranean Fever .
El objetivo biológico de la colchicina es la tubulina, una proteína globular implicada en la composición de los microtúbulos. La colchicina bloquea la polimerización de la tubulina interfiriendo en la migración de los neutrófilos y la desgranulación. Del mismo modo, interfiere en la activación del inflamosoma.
Tratamientos alternativos
Se da la circunstancia de que entre un 5 y un 10% de los pacientes de FMF son resistentes al tratamiento con colchicina (de nuevo, este es mi caso); resistencia que se define clínicamente cuando se sufre al menos una crisis al mes a pesar de mantener un tratamiento diario con 2 mg o más de colchicina.
El mecanismo que provoca esta resistencia no está claro aún, pero un estudio publicado hace diez años mostraba que estos pacientes mantenían una concentración baja del medicamento en las células mononucleares quizás como resultado de un defecto genético aún no identificado.
Durante décadas el único tratamiento conocido era la colchicina, pero los recientes descubrimientos en relación con el inflamosoma así como de la importancia de la interleucina-1β como la citocina más importante involucrada en la patogénesis de la FMF, llevaron a los investigadores a plantear la posibilidad de bloquearla mediante un antagonista .
Actualmente disponemos de tres tratamientos biológicos que actúan como antagonistas para la IL-1β: Anakinra, Rilonacept y Canakinumab. Todos ellos se administran por vía intramuscular aunque difieren en la dosis y la frecuencia de administración. Estos medicamentos son agentes inmunosopresores cuya función es la de bloquear la actividad biológica de la IL-1β producida naturalmente, con lo que ataca la base de la reacción inflamatoria.
Un aspecto que hay que tener muy presente es que, hasta la fecha, no existen ensayos controlados y aleatorizados acerca de la eficacia de estos tratamientos a excepción del Rilonacept, donde se ha comprobado su eficacia en un grupo de 12 pacientes de FMF resistentes a la colchicina . Actualmente se lleva a cabo un ensayo de este tipo (controlado, aleatorizado y de doble ciego) en el Sheba Medical Center de Israel donde se pretende reclutar cincuenta pacientes a los que administrar 100 mg al día de Anakinra o un placebo durante cuatro meses. Hasta abril de este año se contaba con 24 pacientes, 12 de los cuales mostraron una respuesta extraordinaria al medicamento, mientras que los otros 12 continuaron sufriendo ataques. Tendremos que esperar a los resultados definitivos, pero parece que los efectos beneficiosos hay que achacarlos a la medicación real, mientras que los fallos se deberían al placebo.
Conclusiones
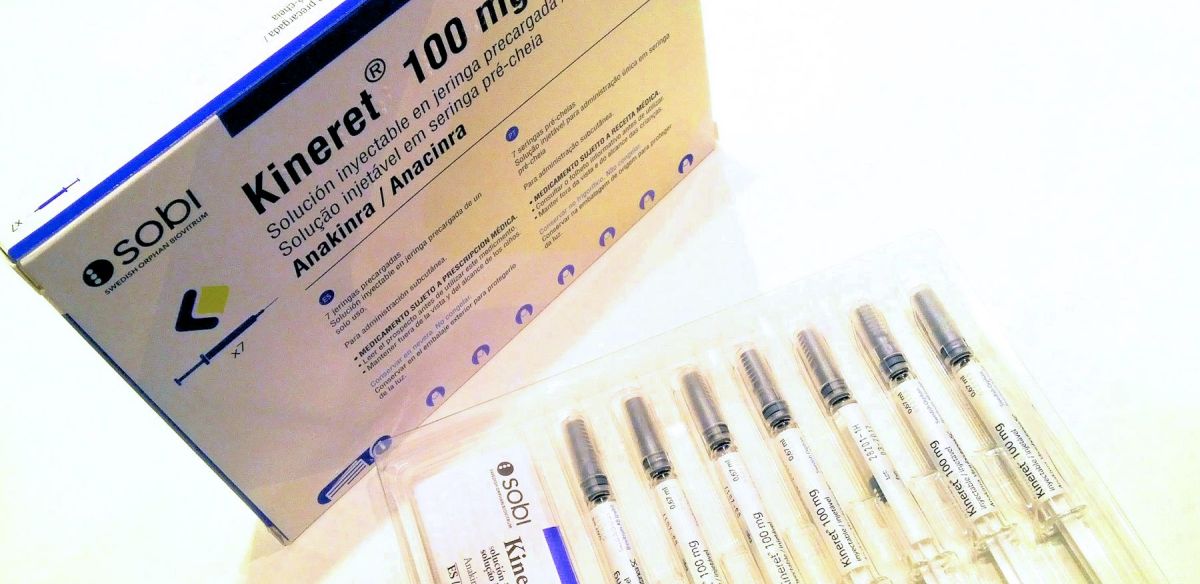
Como he explicado en la charla diré que el pasado día 9 de diciembre comencé el tratamiento con Anakinra (un medicamento bajo el nombre comercial de Kineret) con una dosis inicial de 100 mg al día por vía intravenosa. Aún es pronto para saber si mejorará mi situación, pero lo cierto es que ahora puedo afrontar el futuro con mejores perspectivas.
Para terminar, no puedo dejar de agradecer especialmente la labor del Dr. Manuel Abarca, del Hospital Universitario Virgen de la Victoria de Málaga quien, desde el primer día que acudí a su consulta hace unos meses, se implicó de lleno en mi caso. Su afán por conocer mejor esta enfermedad y buscar una solución para mi caso particular me ha posibilitado el acceso al tratamiento biológico que he comenzado hace poco.

Referencias
Ben-Zvi, I. y Livneh, A. (2014), «Colchicine failure in Familial Mediterranean Fever and potential alternatives: embarking on the anakinra trial«. Isr Med Assoc J, vol. 16, núm. 5, p. 271-273.
Celada, A. (1994), Inmunología básica. Barcelona: Labor, 654 p.
Dinarello, C. A. y van der Meer, J. W. M. (2013), «Treating inflammation by blocking interleukin-1 in humans». Seminars in Immunology, vol. 25, núm. 6, p. 469-484.
Hashkes, P. J., et al. (2012), «Rilonacept for colchicine-resistant or -intolerant Familial Mediterranean Fever: a randomized trial». Ann Intern Med, vol. 157, núm. 8, p. 533-541.
(2014), «Correction: Rilonacept for colchicine-resistant or -intolerant Familial Mediterranean Fever«. Ann Intern Med, vol. 160, núm. 4, p. 291-292.
Lidar, M., et al. (2004), «Colchicine nonresponsiveness in Familial Mediterranean Fever: clinical, genetic, pharmacokinetic, and socioeconomic characterization». Seminars in Arthritis and Rheumatism, vol. 33, núm. 4, p. 273-282.
Matzner, Y., et al. (2000), “Expression of the familial Mediterranean fever gene and activity of the C5a inhibitor in human primary fibroblast cultures“. Blood, vol. 96, núm. 2, p. 727-731. (descarga directa en formato PDF).
Meinzer, U., et al. (2011), «Interleukin-1 targeting drugs in Familial Mediterranean Fever: a case series and a review of the literature«. Seminars in Arthritis and Rheumatism, vol. 41, núm. 2, p. 265-271.
Mitroulis, I., et al. (2008), «Anakinra suppresses Familial Mediterranean Fever crises in a colchicine-resistant patient«. Neth J Med, vol. 66, núm. 11, p. 489-91.
Wang, D. Q. H., et al. (2014), «Familial Mediterranean Fever: from pathogenesis to treatment«. Journal of Genetic Syndromes & Gene Therapy, vol. 5, núm. 5, p. 1-11.
Enlaces
Familial mediterranean fever. OMIM (catálogo de enfermedades genéticas).
Artículos recientes sobre FMF en PubMed.
Notas